Bioconductor 中的 R 包安装方法总结
Bioconductor 是一个基于 R 语言的生物信息软件包,主要用于生物数据的注释、分析、统计、以及可视化 (http://www.bioconductor.org )。
总所周知,Bioconductor 是和 R 版本绑定的,这是为了确保用户不把包安装在错误的版本上。Bioconductor 发行版每年更新两次,它在任何时候都有一个发行版本(release version),对应于 R 的发行版本。此外,Bioconductor 还有一个开发版本(development version),它对应于 R 的开发版本。
R 每年(通常是 4 月中旬)在 ‘x.y.z’ 中发布一个 ‘.y’ 版本,但 Bioconductor 每 6 个月(4 月中旬和 10 月中旬)发布一个 ‘.y’ 版本。
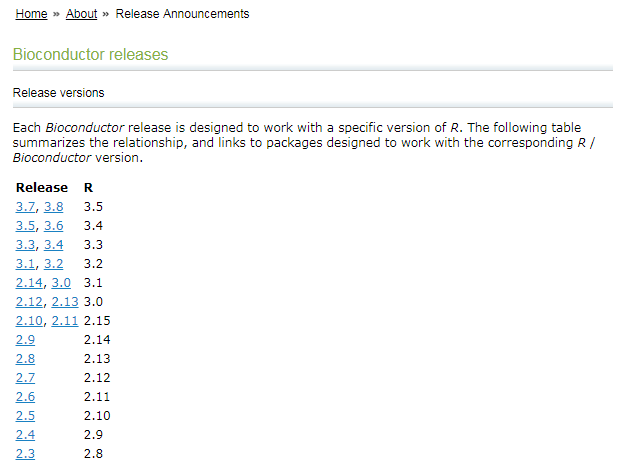
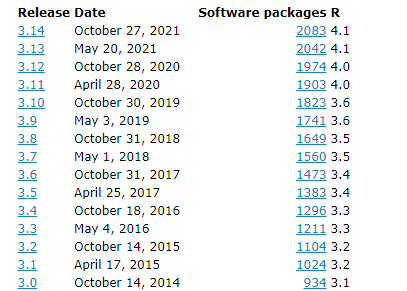
Bioconductor 与 R 各自对应的版本如下:(参考:Bioconductor releases)
biocLite 使用¶
在 R-3.5(Bioconductor-3.7) 前,Bioconductor 都是通过 biocLite 安装相关的 R 包:
source("https://bioconductor.org/biocLite.R")
biocLite(pkg_name)
但是,从 R-3.5(Bioconductor-3.8)起,Bioconductor 更改了 R 包的安装方式:它们通过发布在 CRAN 的 [BiocManager](https://cran.r-project.org/web/packages/BiocManager/index.html) 包来对 Bioconductor 的包进行安装和管理——通过 CRAN 安装 BiocManager,再通过这个包来安装 Bioconductor 的包。
BiocManager 安装与使用¶
1. 镜像,镜像,镜像!¶
重要的事情说三遍!很多安装 CRAN 和 Bioconductor 包的童鞋都会发现自己的包下载不完整,以至于出现各种神奇的报错!所以国内的用户推荐参考下面的用法,设置国内镜像,改善包下载速度慢的问题。
- Bioconductor 镜像
Bioconductor 镜像源配置文件之一是 .Rprofile (Linux 下位于 ~/.Rprofile )。
在文末添加如下语句:
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
打开 R 即可使用该 Bioconductor 镜像源安装 Bioconductor 软件包。
- CRAN 镜像与 R 包安装
R 在线安装包,设置全局镜像(选择中国的镜像),加快安装进度,可以参考以下方法:
#设置清华大学镜像
local({
r <- getOption("repos")
r["CRAN"] <- "http://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)
})
#然后在安装需要的包
install.packages("ggplot2")
或者直接在安装方法中指定 repos,指定国内的镜像地址,安装会快很多:
install.packages("ggplot2",repos="http://mirrors.tuna.tsinghua.edu.cn/CRAN/")
2. 安装 BiocManager 包¶
chooseCRANmirror() # 选择 CRAN 的镜像
install.packages("BiocManager") # 安装 BiocManager 包
3. 安装 Bioconductor 的 R 包¶
BiocManager::install(c("GenomicRanges", "Organism.dplyr"))
4. 查看 Bioconductor 的版本¶
BiocManager::version()
## '3.8'
5. 更新所有已经安装的 R 包¶
BiocManager::install() # 更新到最新版本
6. 旧和意外版本的 R 包¶
当 Bioconductor 的包都来自同一版本时,它们的效果最佳。 使用 valid() 来查看过期(out-of-date)或意外版本(unexpected versions)的 R 包。
BiocManager::valid()
## Warning: 21 packages out-of-date; 2 packages too new
##
## * sessionInfo()
##
## R Under development (unstable) (2018-11-02 r75540)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocStyle_2.11.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.0 bookdown_0.7 digest_0.6.18
## [4] rprojroot_1.3-2 backports_1.1.2 magrittr_1.5
## [7] evaluate_0.12 stringi_1.2.4 rmarkdown_1.10
## [10] tools_3.6.0 stringr_1.3.1 xfun_0.4
## [13] yaml_2.2.0 compiler_3.6.0 BiocManager_1.30.4
## [16] htmltools_0.3.6 knitr_1.20
##
## Bioconductor version '3.9'
##
## * 21 packages out-of-date
## * 2 packages too new
##
## create a valid installation with
##
## BiocManager::install(c(
## "BiocManager", "GenomicDataCommons", "GenomicRanges", "IRanges",
## "RJSONIO", "RcppArmadillo", "S4Vectors", "TCGAbiolinks", "TCGAutils",
## "TMB", "biocViews", "biomaRt", "bumphunter", "curatedMetagenomicData",
## "dimRed", "dplyr", "flowCore", "ggpubr", "ggtree", "lme4", "rcmdcheck",
## "shinyFiles", "tximportData"
## ), update = TRUE, ask = FALSE)
##
## more details: BiocManager::valid()$too_new, BiocManager::valid()$out_of_date
valid() 返回一个对象,可以查询该对象以获取有关无效包的详细信息:
> v <- valid()
Warning message:
6 packages out-of-date; 0 packages too new
> names(v)
[1] "out_of_date" "too_new"
> head(v$out_of_date, 2)
Package LibPath
bit "bit" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
ff "ff" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
Installed Built ReposVer Repository
bit "1.1-12" "3.5.0" "1.1-13" "https://cran.rstudio.com/src/contrib"
ff "2.2-13" "3.5.0" "2.2-14" "https://cran.rstudio.com/src/contrib"
>
7. 适用的 R 包¶
可以使用 available() 发现适用于我们的 Bioconductor 版本的软件包;第一个参数是可用于根据正则表达式过滤包名称,例如,可用于 Homo sapiens 的 ‘BSgenome’ 包:
avail <- BiocManager::available()
length(avail)
## [1] 16261
BiocManager::available("BSgenome.Hsapiens")
## [1] "BSgenome.Hsapiens.1000genomes.hs37d5"
## [2] "BSgenome.Hsapiens.NCBI.GRCh38"
## [3] "BSgenome.Hsapiens.UCSC.hg17"
## [4] "BSgenome.Hsapiens.UCSC.hg17.masked"
## [5] "BSgenome.Hsapiens.UCSC.hg18"
## [6] "BSgenome.Hsapiens.UCSC.hg18.masked"
## [7] "BSgenome.Hsapiens.UCSC.hg19"
## [8] "BSgenome.Hsapiens.UCSC.hg19.masked"
## [9] "BSgenome.Hsapiens.UCSC.hg38"
## [10] "BSgenome.Hsapiens.UCSC.hg38.masked"
安装旧版本的 Bioconductor R 包¶
R≥3.5,Bioconductor≥3.7¶
可以使用 BiocManager 安装相关与版本匹配的 R 包。或者通过源码的方式安装旧版本 R 包。
R<3.5,Bioconductor<3.7¶
那么使用 3.5 以下 R 版本的用户是继续使用 biocLite,还是 BiocManager,还是其他的方法安装匹配相关版本的 R 包呢?
**首先,**对于 R < 3.5.0,如果 biocLite 或者 BiocManager 可以安装,则优先使用 biocLite 或者 BiocManager 去安装。
**其次,**对于 R < 3.5.0, 如果 biocLite 和 BiocManager 都无法安装对应版本的 R 包,可以参考下面的方法。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
......
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)
> chooseCRANmirror()
> install.packages("BiocManager")
Warning message:
package ‘BiocManager’ is not available (for R version 3.4.3)
>
这时候,Bioconductor 推荐使用以下命令安装相应的 R 包。
source("https://bioconductor.org/biocLite.R")
BiocInstaller::biocLite(c("GenomicFeatures", "AnnotationDbi"))
安装新版本的 Bioconductor R 包¶
Bioconductor 是与特定版本的 R 绑定的,正常来说当 Bioconductor 的包都来自同一版本时,它们的效果最佳。
Bioconductor versions are associated with specific R versions, as summarized here. Attempting to install a version of Bioconductor that is not supported by the version of R in use leads to an error; using the most recent version of Bioconductor may require installing a new version of R.
From:https://cran.r-project.org/web/packages/BiocManager/vignettes/BiocManager.html
所以,当有些 R 包是基于高版本的 Bioconductor 开发的,在低版本的 Bioconductor/R 中直接执行 BiocManager::install("package"),安装得到的 package 版本默认是与当前版本 Bioconductor/R 相匹配的,而并非是最新的版本。
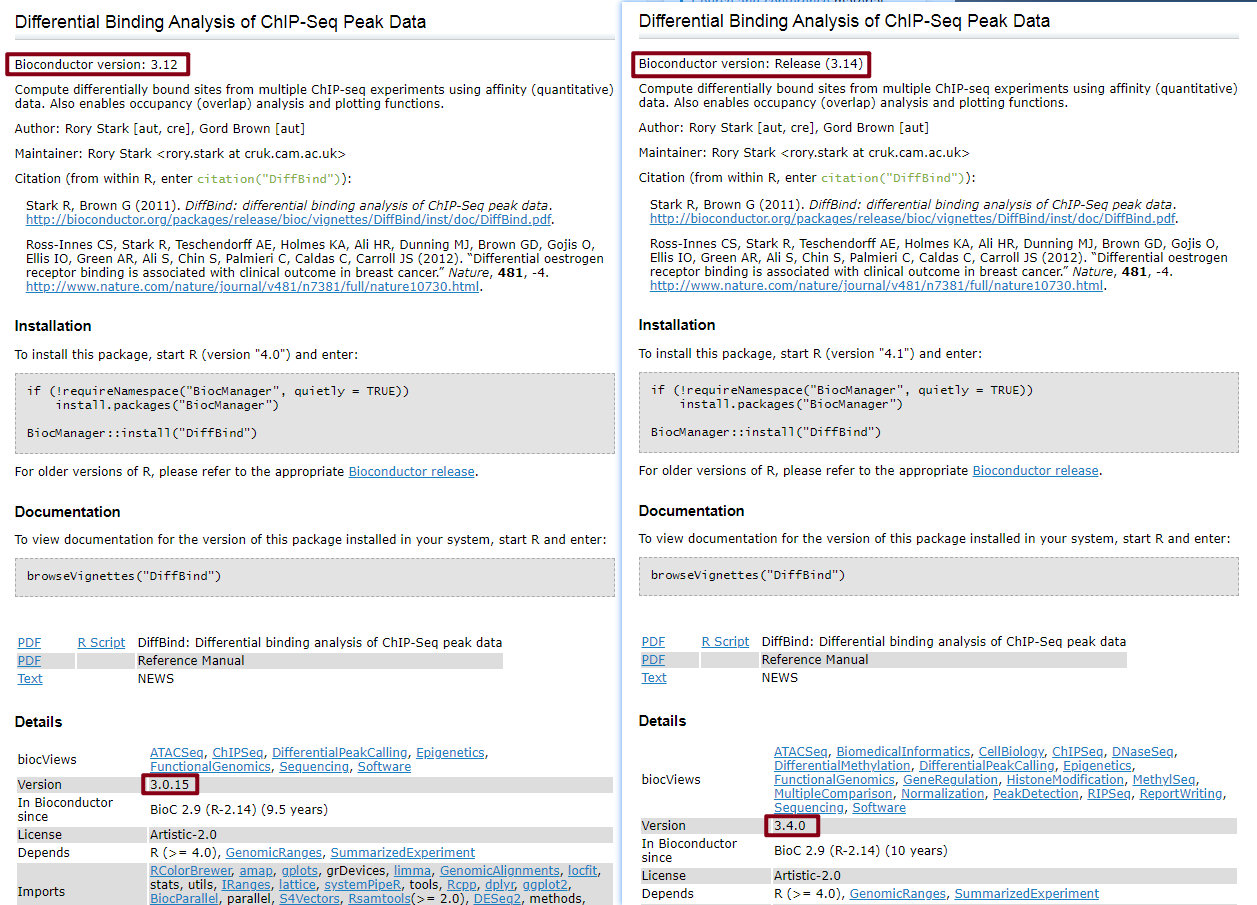 以 DiffBind 包为例,DiffBind==3.4.0 是基于 Bioconductor==3.14(对应 R-4.1)开发的;我们在 Bioconductor==3.13(对应 R-4.0)中执行
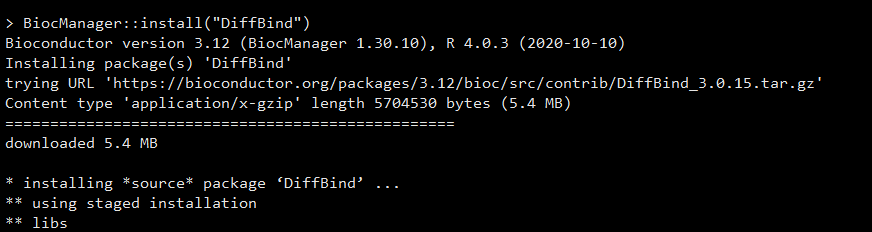
以 DiffBind 包为例,DiffBind==3.4.0 是基于 Bioconductor==3.14(对应 R-4.1)开发的;我们在 Bioconductor==3.13(对应 R-4.0)中执行 BiocManager::install("DiffBind"),默认安装的是 DiffBind==3.0.15!
1. 源码方式安装¶
如果想要在 Bioconductor==3.13(对应 R-4.0)中安装 DiffBind==3.4.0,可以直接通过源码包的方式安装:
> packageurl <- "http://bioconductor.org/packages/release/bioc/src/contrib/DiffBind_3.4.0.tar.gz"
> install.packages(packageurl, repos=NULL, type="source")
2. BiocInstaller 安装¶
下面,我们以在 R-3.4(Bioconductor==3.6)中安装最新版本的 clusterProfiler 为例。
在 Aanconda2 环境 R==3.4.3 中安装 clusterProfiler,发现 package ‘clusterProfile’ is not available (for R version 3.4.3)。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfile’
Old packages: 'ade4', 'ape', 'backports', 'caret', ......
Update all/some/none? [a/s/n]: n
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)
使用 BiocInstaller 安装 clusterProfiler:
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> BiocInstaller::biocLite("clusterProfiler")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfiler’
trying URL 'https://bioconductor.org/packages/3.6/bioc/src/contrib/clusterProfiler_3.6.0.tar.gz'
Content type 'application/x-gzip' length 4478098 bytes (4.3 MB)
==================================================
downloaded 4.3 MB
* installing *source* package ‘clusterProfiler’ ...
** R
** data
** inst
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
** building package indices
** installing vignettes
** testing if installed package can be loaded
* DONE (clusterProfiler)
> packageVersion('clusterProfiler')
[1] ‘3.6.0’
install.packages 一站式方案¶
用 install.packages 来安装 CRAN 和 Bioconductor 所有的包!这是来自于 Y 叔 2018-09-25 在公众号发表的《不用 biocLite 安装 Bioconductor 包》介绍的方法。这里截取部分内容介绍一下。
用 install.packages 来安装 CRAN 和 Bioconductor 所有的包,你要做的很简单,在 \~/.Rprofile 里加入以下两行内容。
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
utils::setRepositories(ind=1:2)
第一行,使用国内的镜像,我这里用的是清华大学的,第二行,设定
install.packages从 CRAN 和 Bioconductor 中搜索包,其实你还可以让它支持比如 R-Forge 以及各种第三方的仓库。然后你就可以愉快地使用
install.packages来安装 Bioconductor 包了。
安装体积比较大的 R 包¶
安装 CRAN 或者 Bioconductor 中一些体积比较大的 R 包,如果网络不太好,经常可能会出现包下载不完(Timeout of 60 seconds was reached),从而导致无法正常安装。
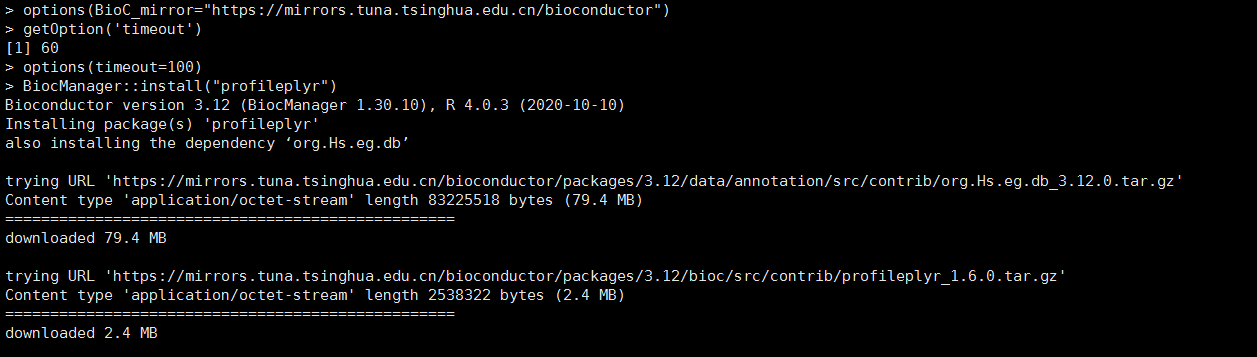
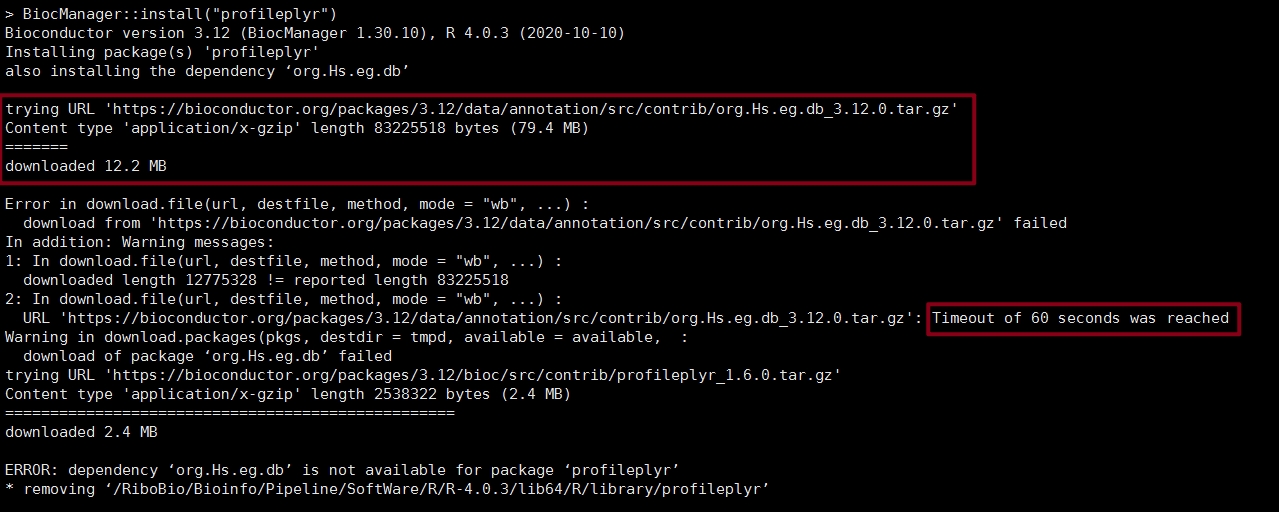 参考 How do i set a timeout for utils::download.file() in R - Stack Overflow,增加 timeout 时长的同时使用国内的镜像进行加速:
参考 How do i set a timeout for utils::download.file() in R - Stack Overflow,增加 timeout 时长的同时使用国内的镜像进行加速:
getOption('timeout')
# [1] 60
options(timeout=100)
以上,就是 Bioconductor R 包安装和使用的全部内容,希望对大家有所帮助。
参考资料¶
- omicsgene,《R 语言包安装方法,设置国内镜像加快安装速度》,OmicsClass 组学大讲堂问答社区
- Y 叔叔,《不用 biocLite 安装 Bioconductor 包》,”biobable”公众号